
Transfer of Cardiac Mitochondria Improves the Therapeutic Efficacy of Mesenchymal Stem Cells in a Preclinical Model of Ischemic Heart Disease
 , , , , and
, , , , and 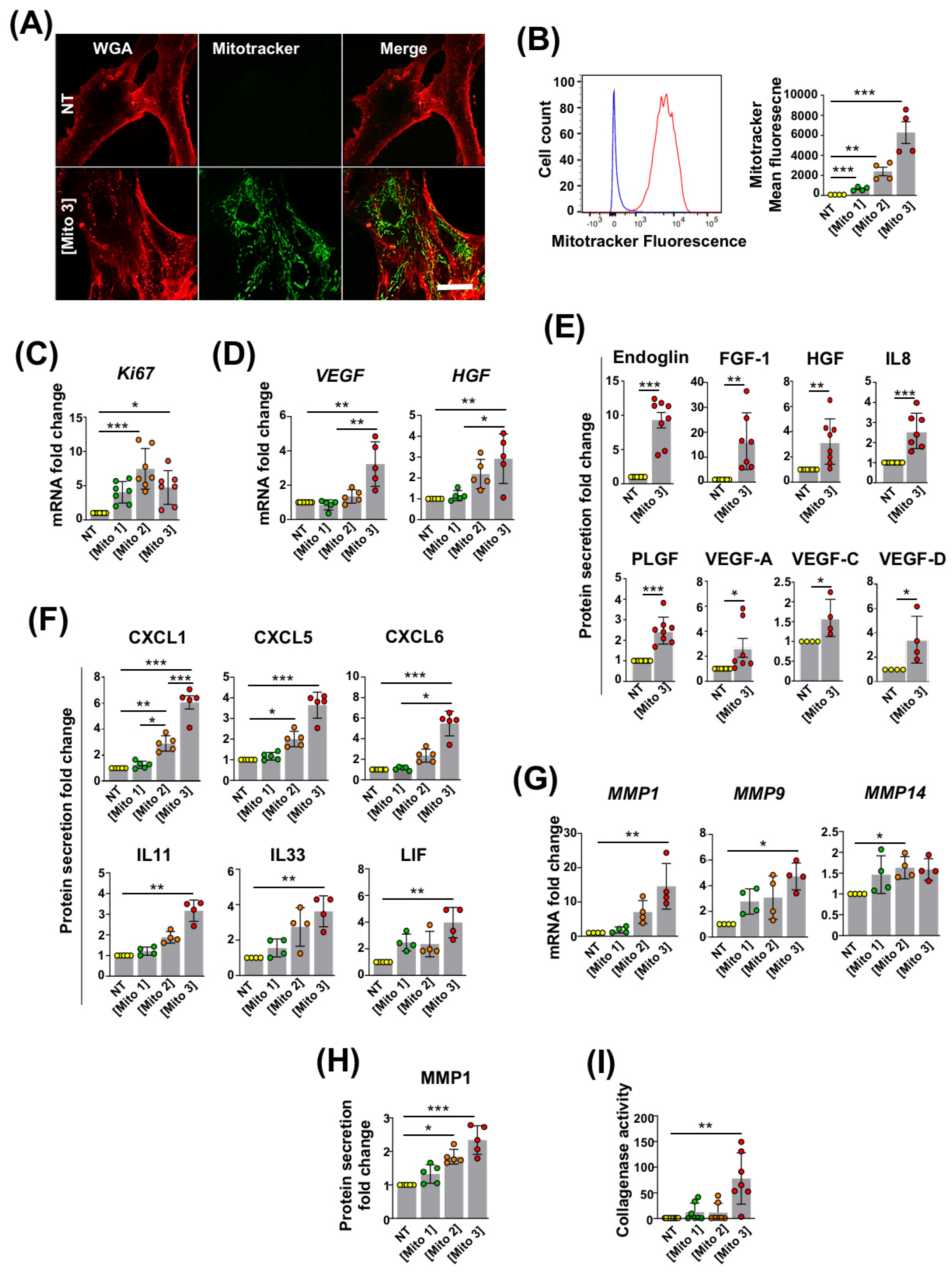{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Cardiac Mitochondria Isolation and Transfer to MSCs
2.3. Inhibition of Cardiac Mitochondria Transfer to MSCs
2.4. Inhibition of MSC Autophagy
2.5. Cardiac Mitochondria-Derived ROS Analysis
2.6. Cardiac Mitochondria Mitophagy
2.7. Transmission Electron Microscopy
2.8. Real-Time PCR Assays
2.9. Collection of MSC Conditioned Media
2.10. ELISAs
2.11. Luminex Assays
2.12. Collagenase Enzymatic Activity
2.13. Murine Model of Myocardial Infarction and Cell Grafting
2.14. Echocardiography and Speckle Tracking Analysis
2.15. Photoacoustic Imaging
2.16. Statistical Analysis
3. Results
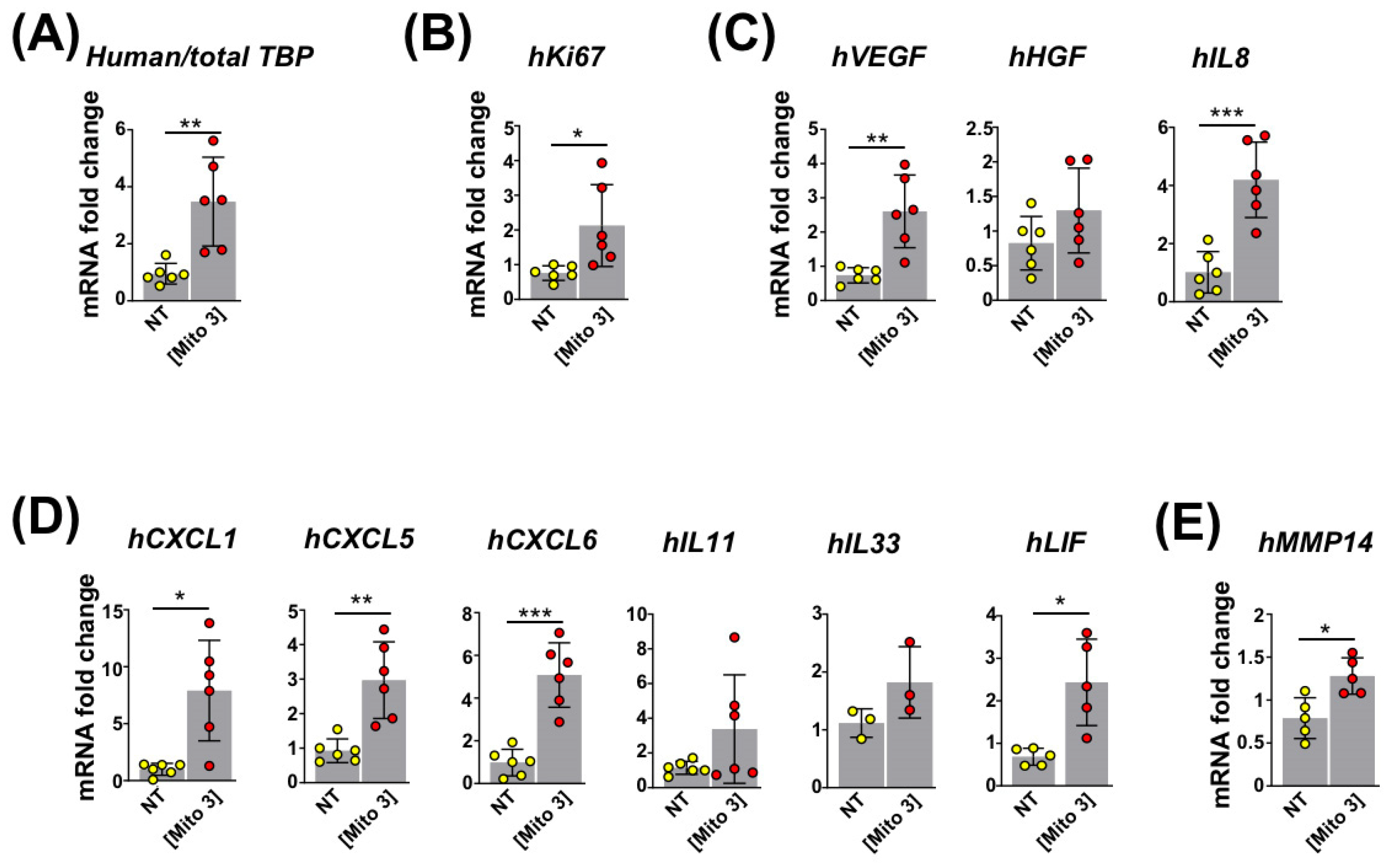
3.1. Transfer of Cardiac Mitochondria Stimulates Proliferation and the Regenerative Properties of Recipient MSCs
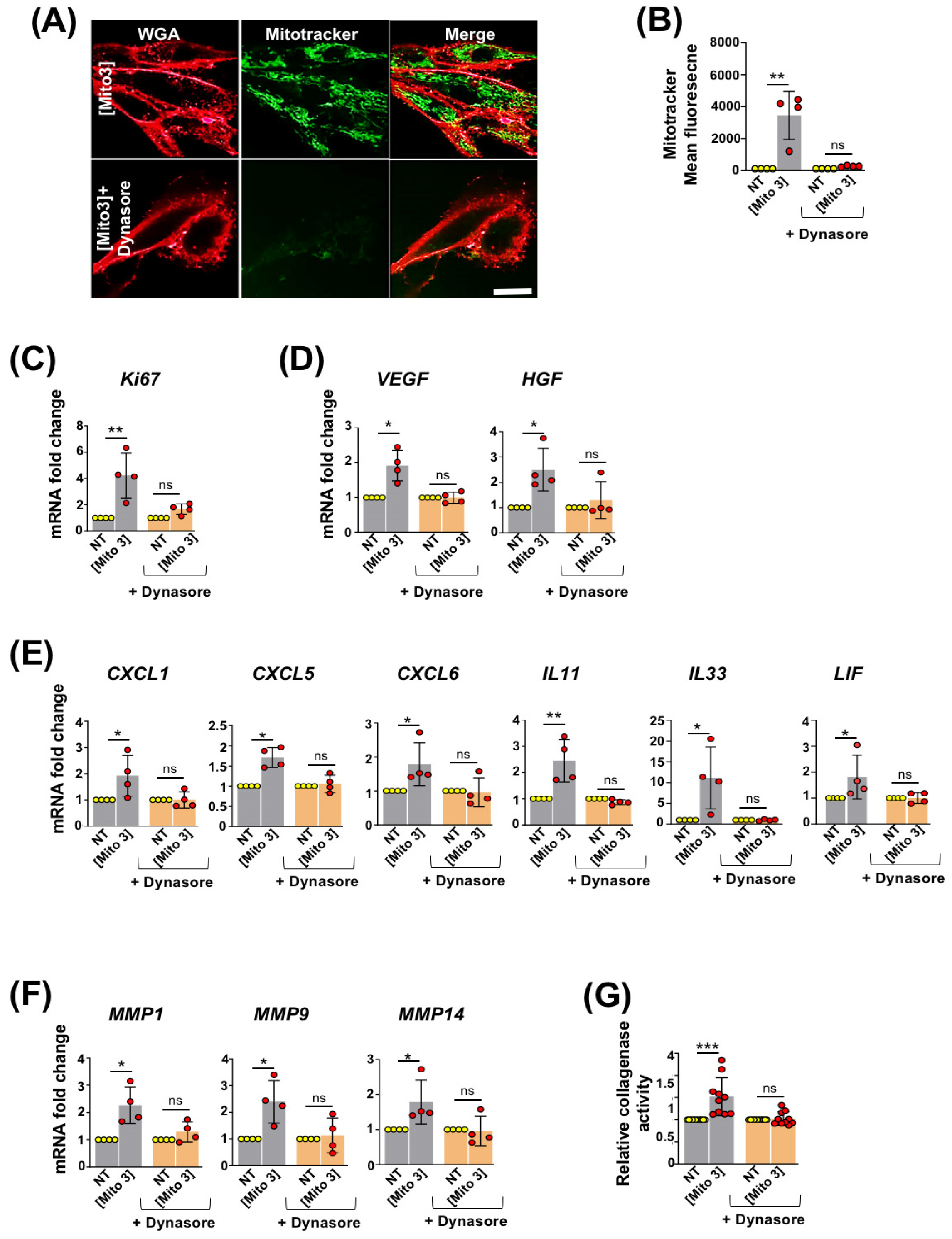
3.2. Free Cardiac Mitochondria Are Transferred to MSCs via Dynamin-Dependent, Clathrin-Mediated Endocytosis
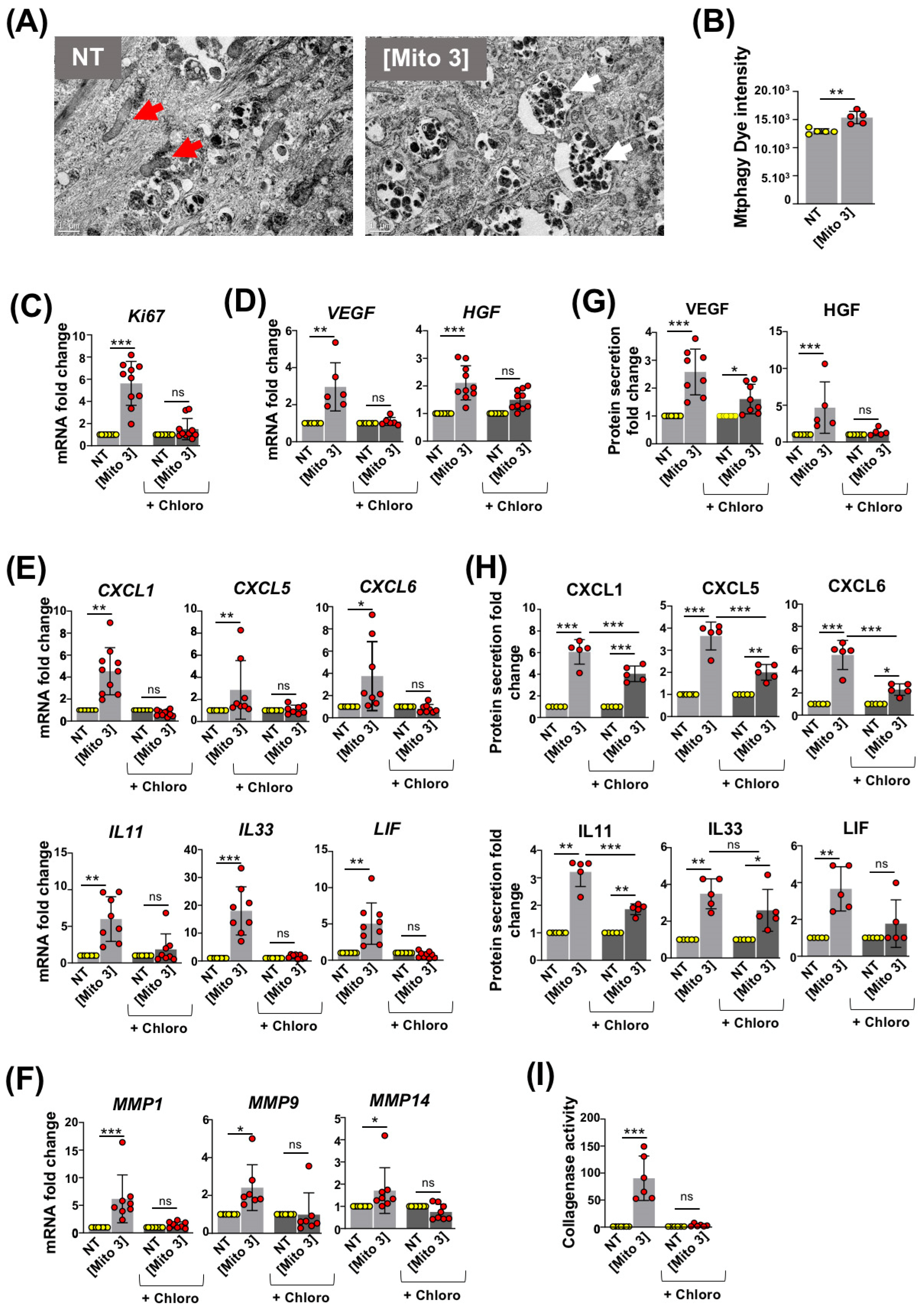
3.3. Degradation of Cardiac Mitochondria Transferred to MSCs Is Necessary to Trigger Their Therapeutic Potential
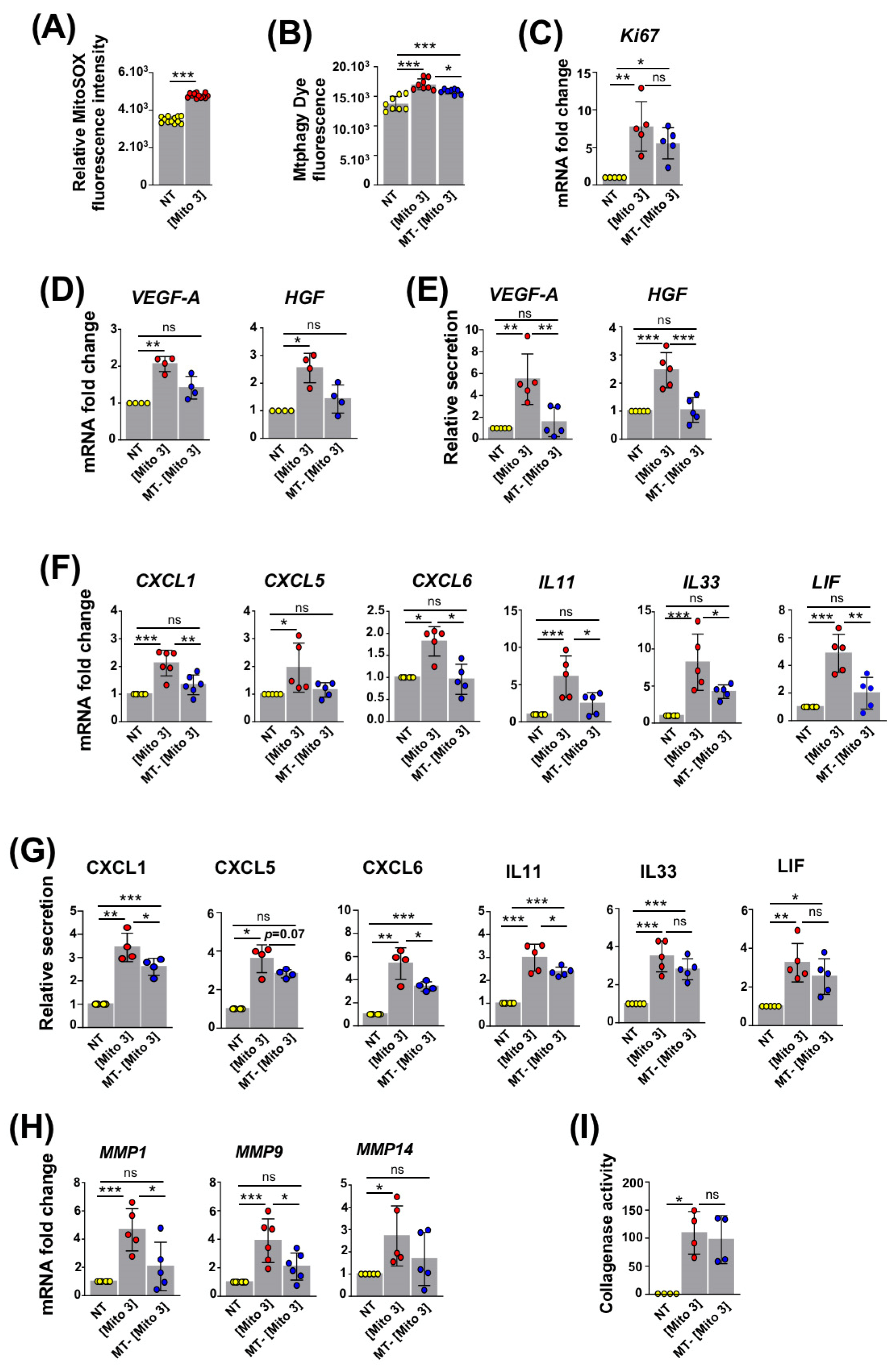
3.4. ROS Produced by Cardiac Mitochondria Trigger Autophagy and Activation of Recipient MSCs
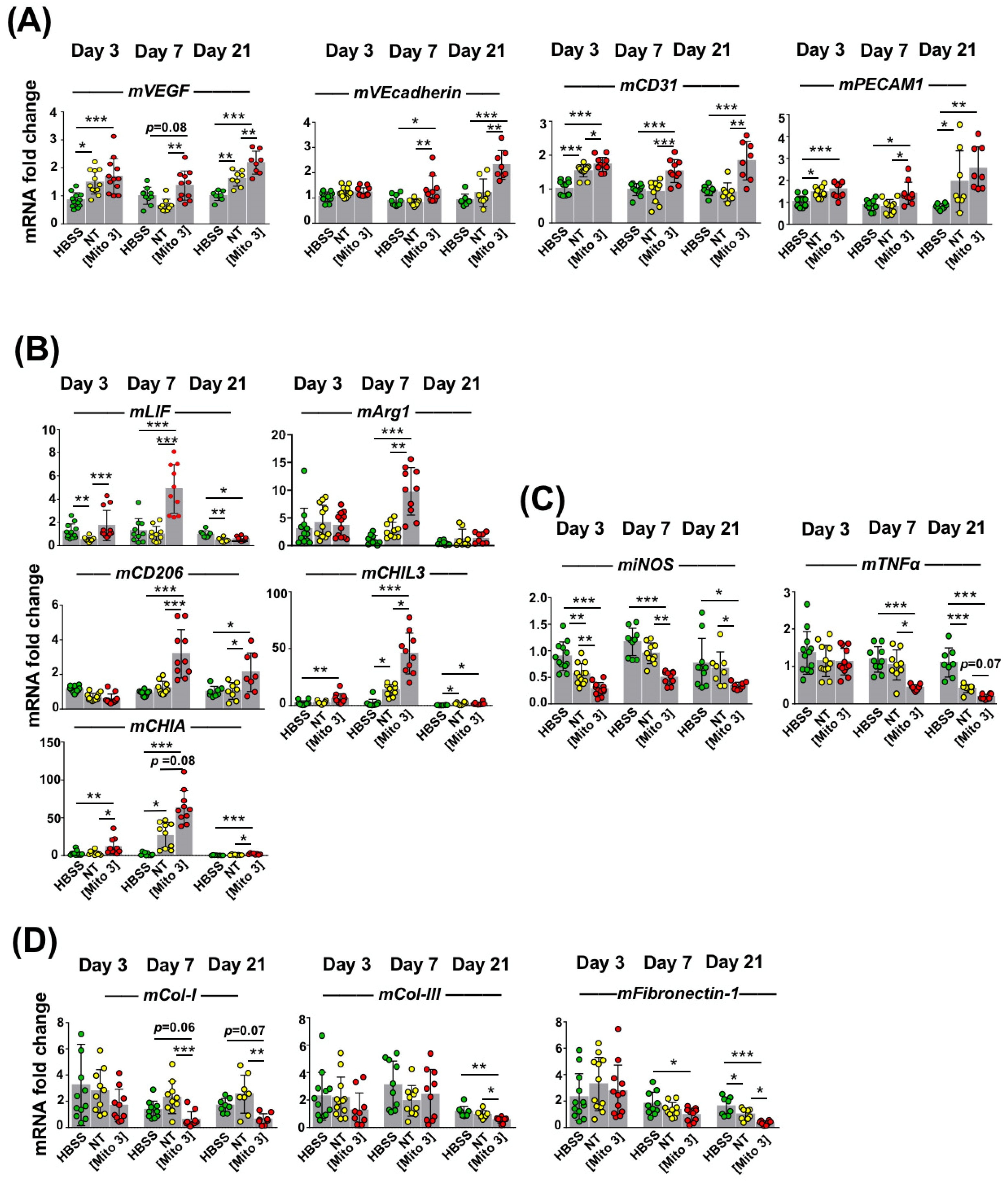
3.5. Transfer of Cardiac Mitochondria Enhances the Therapeutic Potential of Transplanted MSCs in a Mouse Model of Ischemic Cardiomyopathy
3.6. Mitochondria-Preconditioned MSCs Improve Post-MI LV Function and Perfusion
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leong, D.P.; Joseph, P.G.; McKee, M.; Anand, S.S.; Teo, K.K.; Schwalm, J.D.; Salim, Y. Reducing the Global Burden of Cardiovascular Disease, Part 2: Prevention and Treatment of Cardiovascular Disease. Circ. Res. 2017, 121, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Agbulut, O.; Mazo, M.; Bressolle, C.; Gutierrez, M.; Azarnoush, K.; Sabbah, L.; Niederlander, N.; Abizanda, G.; Andreu, E.J.; Pelacho, B.; et al. Can bone marrow-derived multipotent adult progenitor cells regenerate infarcted myocardium? Cardiovasc. Res. 2006, 72, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, H.; Planat-Benard, V.; Bel, A.; Puymirat, E.; Geha, R.; Pidial, L.; Nematalla, H.; Bellamy, V.; Bouaziz, P.; Peyrard, S.; et al. Epicardial adipose stem cell sheets results in greater post-infarction survival than intramyocardial injections. Cardiovasc. Res. 2011, 91, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, H.; Planat-Benard, V.; Bel, A.; Puymirat, E.; Geha, R.; Pidial, L.; Nematalla, H.; Bellamy, V.; Bouaziz, P.; Peyrard, S.; et al. Long-term functional benefits of epicardial patches as cell carriers. Cell Transplant. 2014, 23, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Müller, P.; Lemcke, H.; David, R. Stem Cell Therapy in Heart Diseases—Cell Types, Mechanisms and Improvement Strategies. Cell. Physiol. Biochem. 2018, 48, 607–655. [Google Scholar] [CrossRef]
- Razeghian-Jahromi, I.; Matta, A.G.; Canitrot, R.; Zibaeenezhad, M.J.; Razmkhah, M.; Safari, A.; Nader, V.; Roncalli, J. Surfing the clinical trials of mesenchymal stem cell therapy in ischemic cardiomyopathy. Stem. Cell Res. Ther. 2021, 12, 361. [Google Scholar] [CrossRef]
- Ferraro, F.; Celso, C.L.; Scadden, D. Adult Stem Cells and Their Niches. Adv. Exp. Med. Biol. 2010, 695, 155–168. [Google Scholar]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell. Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef]
- Wei, Q.; Frenette, P.S. Niches for Hematopoietic Stem Cells and Their Progeny. Immunity 2018, 48, 632–648. [Google Scholar] [CrossRef]
- Figeac, F.; Lesault, P.F.; le Coz, O.; Damy, T.; Souktani, R.; Trébeau, C.; Schmitt, A.; Ribot, J.; Mounier, R.; Guguin, A.; et al. Nanotubular Crosstalk with Distressed Cardiomyocytes Stimulates the Paracrine Repair Function of Mesenchymal Stem Cells: Cardiomyocyte Crosstalk Improves Stem Cell Healing. Stem Cells 2014, 32, 216–230. [Google Scholar] [CrossRef]
- Li, F.; Huang, Q.; Chen, J.; Peng, Y.; Roop, D.; Bedford, J.S.; Li, C.-Y. Apoptotic Cells Activate the “Phoenix Rising” Pathway to Promote Wound Healing and Tissue Regeneration. Sci. Signal. 2010, 3, ra13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, A.M.; Nakhle, J.; Griessinger, E.; Vignais, M.L. Intercellular mitochondria trafficking highlighting the dual role of mesenchymal stem cells as both sensors and rescuers of tissue injury. Cell Cycle 2018, 17, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Vallabhaneni, K.C.; Haller, H.; Dumler, I. Vascular Smooth Muscle Cells Initiate Proliferation of Mesenchymal Stem Cells by Mitochondrial Transfer via Tunneling Nanotubes. Stem Cells Dev. 2012, 21, 3104–3113. [Google Scholar] [CrossRef] [PubMed]
- Koyanagi, M.; Brandes, R.P.; Haendeler, J.; Zeiher, A.M.; Dimmeler, S. Cell-to-Cell Connection of Endothelial Progenitor Cells with Cardiac Myocytes by Nanotubes. Circ. Res. 2005, 96, 1039–1041. [Google Scholar] [CrossRef]
- Plotnikov, E.Y.; Khryapenkova, T.G.; Galkina, S.I.; Sukhikh, G.T.; Zorov, D.B. Cytoplasm and organelle transfer between mesenchymal multipotent stromal cells and renal tubular cells in co-culture. Exp. Cell Res. 2010, 316, 2447–2455. [Google Scholar] [CrossRef]
- Mahrouf-Yorgov, M.; Augeul, L.; Da Silva, C.C.; Jourdan, M.; Rigolet, M.; Manin, S.; Ferrera, R.; Ovize, M.; Henry, A.; Guguin, A.; et al. Mesenchymal stem cells sense mitochondria released from damaged cells as danger signals to activate their rescue properties. Cell Death Differ. 2017, 24, 1224–1238. [Google Scholar] [CrossRef]
- Levoux, J.; Prola, A.; Lafuste, P.; Gervais, M.; Chevallier, N.; Koumaiha, Z.; Kefi, K.; Braud, L.; Schmitt, A.; Yacia, A.; et al. Platelets Facilitate the Wound-Healing Capability of Mesenchymal Stem Cells by Mitochondrial Transfer and Metabolic Reprogramming. Cell Metab. 2021, 33, 283–299.e9. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab. 2017, 26, 9–48. [Google Scholar] [CrossRef]
- Yan, W.; Diao, S.; Fan, Z. The role and mechanism of mitochondrial functions and energy metabolism in the function regulation of the mesenchymal stem cells. Stem Cell Res. Ther. 2021, 12, 140. [Google Scholar] [CrossRef]
- Doménech, E.; Maestre, C.; Esteban-Martínez, L.; Partida, D.; Pascual, R.; Fernández-Miranda, G.; Seco, E.; Campos-Olivas, R.; Pérez, M.; Megias, D.; et al. AMPK and PFKFB3 mediate glycolysis and survival in response to mitophagy during mitotic arrest. Nat. Cell. Biol. 2015, 17, 1304–1316. [Google Scholar] [CrossRef]
- Esteban-Martínez, L.; Sierra-Filardi, E.; Boya, P. Mitophagy, metabolism, and cell fate. Mol. Cell. Oncol. 2017, 4, e1353854. [Google Scholar] [CrossRef]
- Esteban-Martínez, L.; Sierra-Filardi, E.; McGreal, R.S.; Salazar-Roa, M.; Mariño, G.; Seco, E.; Durand, S.; Enot, D.; Graña, O.; Malumbres, M.; et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J. 2017, 36, 1688–1706. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.P.; Birbrair, A.; Bhutia, S.K. Mitophagy-driven metabolic switch reprograms stem cell fate. Cell. Mol. Life Sci. 2019, 76, 27–43. [Google Scholar] [CrossRef]
- Rodriguez, A.M.; Pisani, D.; Dechesne, C.A.; Turc-Carel, C.; Kurzenne, J.Y.; Wdziekonski, B.; Villageois, A.; Bagnis, C.; Breittmayer, J.-P.; Groux, H.; et al. Transplantation of a multipotent cell population from human adipose tissue induces dystrophin expression in the immunocompetent mdx mouse. J. Exp. Med. 2005, 201, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Maayah, Z.H.; Elshenawy, O.H.; Althurwi, H.N.; Abdelhamid, G.; El-Kadi, A.O.S. Human fetal ventricular cardiomyocyte, RL-14 cell line, is a promising model to study drug metabolizing enzymes and their associated arachidonic acid metabolites. J. Pharmacol. Toxicol. Methods 2015, 71, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Zacchigna, S.; Paldino, A.; Falcão-Pires, I.; Daskalopoulos, E.P.; dal Ferro, M.; Vodret, S.; Lesizza, P.; Cannatà, A.; Miranda-Silva, D.; Lourenço, A.P.; et al. Towards standardization of echocardiography for the evaluation of left ventricular function in adult rodents: A position paper of the ESC Working Group on Myocardial Function. Cardiovasc. Res. 2021, 117, 43–59. [Google Scholar] [CrossRef]
- David, H.; Ughetto, A.; Gaudard, P.; Plawecki, M.; Paiyabhroma, N.; Zub, E.; Colson, P.; Richard, S.; Marchi, N.; Sicard, P.; et al. Experimental Myocardial Infarction Elicits Time-Dependent Patterns of Vascular Hypoxia in Peripheral Organs and in the Brain. Front. Cardiovasc. Med. 2021, 7, 615507. [Google Scholar] [CrossRef]
- Schofield, J.H.; Schafer, Z.T. Mitochondrial Reactive Oxygen Species and Mitophagy: A Complex and Nuanced Relationship. Antioxid. Redox Signal. 2021, 34, 517–530. [Google Scholar] [CrossRef]
- Fauconnier, J.; Andersson, D.C.; Zhang, S.J.; Lanner, J.T.; Wibom, R.; Katz, A.; Bruton, J.D.; Westerblad, H. Effects of Palmitate on Ca2+ Handling in Adult Control and ob/ob Cardiomyocytes: Impact of Mitochondrial Reactive Oxygen Species. Diabetes 2007, 56, 1136–1142. [Google Scholar] [CrossRef]
- Hackfort, B.T.; Chalise, U.; Daseke, M.J.; Lindsey, M.L. Myocardial Oxygen Saturation Measured by Photoacoustic EKV Imaging. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Yamada, Y.; Ito, M.; Arai, M.; Hibino, M.; Tsujioka, T.; Harashima, H. Challenges in Promoting Mitochondrial Transplantation Therapy. Int. J. Mol. Sci. 2020, 21, 6365. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Kumar, M.; Singh, S.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [PubMed]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J.; et al. Mitochondrial transfer from bone-marrow–derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Doulamis, I.P.; Guariento, A.; Duignan, T.; Orfany, A.; Kido, T.; Zurakowski, D.; del Nido, P.J.; McCully, J.D. Mitochondrial transplantation for myocardial protection in diabetic hearts. Eur. J. Cardio Thorac. Surg. 2020, 57, 836–845. [Google Scholar] [CrossRef]
- Kaza, A.K.; Wamala, I.; Friehs, I.; Kuebler, J.D.; Rathod, R.H.; Berra, I.; Ericsson, M.; Yao, R.; Thedsanamoorthy, J.K.; Zurakowski, D.; et al. Myocardial rescue with autologous mitochondrial transplantation in a porcine model of ischemia/reperfusion. J. Thorac. Cardiovasc. Surg. 2017, 153, 934–943. [Google Scholar] [CrossRef]
- Masuzawa, A.; Black, K.M.; Pacak, C.A.; Ericsson, M.; Barnett, R.J.; Drumm, C.; Seth, P.; Bloch, D.B.; Levitsky, S.; Cowan, D.B.; et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am. J. Physiol. -Heart Circ. Physiol. 2013, 304, H966–H982. [Google Scholar] [CrossRef]
- McCully, J.D.; Cowan, D.B.; Pacak, C.A.; Toumpoulis, I.K.; Dayalan, H.; Levitsky, S. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H94–H105. [Google Scholar] [CrossRef]
- Emani, S.M.; Piekarski, B.L.; Harrild, D.; del Nido, P.J.; McCully, J.D. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2017, 154, 286–289. [Google Scholar] [CrossRef]
- Liao, P.C.; Bergamini, C.; Fato, R.; Pon, L.A.; Pallotti, F. Isolation of mitochondria from cells and tissues. Methods Cell Biol. 2020, 155, 3–31. [Google Scholar] [PubMed]
- Babenko, V.A.; Silachev, D.N.; Popkov, V.A.; Zorova, L.D.; Pevzner, I.B.; Plotnikov, E.Y.; Sukhikh, G.T.; Zorov, D.B. Miro1 Enhances Mitochondria Transfer from Multipotent Mesenchymal Stem Cells (MMSC) to Neural Cells and Improves the Efficacy of Cell Recovery. Molecules 2018, 23, 687. [Google Scholar] [CrossRef] [PubMed]
- Kanelidis, A.J.; Premer, C.; Lopez, J.; Balkan, W.; Hare, J.M. Route of Delivery Modulates the Efficacy of Mesenchymal Stem Cell Therapy for Myocardial Infarction: A Meta-Analysis of Preclinical Studies and Clinical Trials. Circ. Res. 2017, 120, 1139–1150. [Google Scholar] [CrossRef]
- Guo, Y.; Yu, Y.; Hu, S.; Chen, Y.; Shen, Z. The therapeutic potential of mesenchymal stem cells for cardiovascular diseases. Cell Death Dis. 2020, 11, 349. [Google Scholar] [CrossRef]
- Khan, K.; Gasbarrino, K.; Mahmoud, I.; Dufresne, L.; Daskalopoulou, S.S.; Schwertani, A.; Cecere, R. Bioactive Scaffolds in Stem Cell-Based Therapies for Myocardial Infarction: A Systematic Review and Meta-Analysis of Preclinical Trials. Stem Cell Rev. Rep. 2021, 8, 2104–2136. [Google Scholar] [CrossRef] [PubMed]
- Golpanian, S.; Schulman, I.H.; Ebert, R.F.; Heldman, A.W.; DiFede, D.L.; Yang, P.C.; Wu, J.C.; Bolli, R.; Perin, E.C.; Moyé, L.; et al. Concise Review: Review and Perspective of Cell Dosage and Routes of Administration from Preclinical and Clinical Studies of Stem Cell Therapy for Heart Disease. Stem Cells Transl. Med. 2016, 5, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar] [CrossRef]
- Lian, Q.; Zhang, Y.; Liang, X.; Gao, F.; Tse, H.F. Directed Differentiation of Human-Induced Pluripotent Stem Cells to Mesenchymal Stem Cells. Methods Mol. Biol. 2016, 1416, 289–298. [Google Scholar] [PubMed]
- Bloor, A.J.C.; Patel, A.; Griffin, J.E.; Gilleece, M.H.; Radia, R.; Yeung, D.T.; Drier, D.; Larson, L.S.; Uenishi, G.I.; Hei, D.; et al. Production, safety and efficacy of iPSC-derived mesenchymal stromal cells in acute steroid-resistant graft versus host disease: A phase I, multicenter, open-label, dose-escalation study. Nat. Med. 2020, 26, 1720–1725. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vignais, M.-L.; Levoux, J.; Sicard, P.; Khattar, K.; Lozza, C.; Gervais, M.; Mezhoud, S.; Nakhle, J.; Relaix, F.; Agbulut, O.; et al. Transfer of Cardiac Mitochondria Improves the Therapeutic Efficacy of Mesenchymal Stem Cells in a Preclinical Model of Ischemic Heart Disease. Cells 2023, 12, 582. https://doi.org/10.3390/cells12040582
Vignais M-L, Levoux J, Sicard P, Khattar K, Lozza C, Gervais M, Mezhoud S, Nakhle J, Relaix F, Agbulut O, et al. Transfer of Cardiac Mitochondria Improves the Therapeutic Efficacy of Mesenchymal Stem Cells in a Preclinical Model of Ischemic Heart Disease. Cells. 2023; 12(4):582. https://doi.org/10.3390/cells12040582
Chicago/Turabian StyleVignais, Marie-Luce, Jennyfer Levoux, Pierre Sicard, Khattar Khattar, Catherine Lozza, Marianne Gervais, Safia Mezhoud, Jean Nakhle, Frederic Relaix, Onnik Agbulut, and et al. 2023. "Transfer of Cardiac Mitochondria Improves the Therapeutic Efficacy of Mesenchymal Stem Cells in a Preclinical Model of Ischemic Heart Disease" Cells 12, no. 4: 582. https://doi.org/10.3390/cells12040582